
由于大多数微生物的不可培养性,宏基因组学现已成为研究微生物群落最有效的手段,而利用宏基因组组装基因组(MAGs)的有效性主要取决于微生物群体的复杂性、目标菌群的丰度以及测序的深度。目前利用Hi-C技术开展宏基因组研究已成为宏基因组组装的新利器。今天小编就带大家一起来看看Hi-C和宏基因组是如何完美结合的吧!

数据来源: 从基因组分类数据库(GTDB)中随机选择63个高质量的细菌基因组,设计了一个模拟的人类肠道微生物组。选择标准:contig count<200,完整性>98%,总gap<500bp,共有223个满足此标准的候选基因组;
真实肠道微生物样本采用Illumina HiSeqX Ten PE150测序,宏基因组数据量为74.6G,两个Hi-C文库数据量分别为1.3G (SRR6131122)和1.2G(SRR6131124)。
数据分析:宏基因组数据使用宏基因组散弹枪模拟器MetaART产生18.2M pairs(250X);MetaART包含短读数模拟器art-illumina(V2.5.1) Hi-C数据由两种不同的四碱基酶(NEB:MluCI 和 Sau3Al)共产生200M read pairs。
FastANI (v1.0)计算223个候选基因组序列之间的成对平均核苷酸一致性(ANI) ;采用BBTools (v37.25)对初始读集按因子进行连续缩减采样,初始的Hi-C读集减少了4次,共5个不同深度(200M,100M,50M,25M,12.5M pairs)。Hi-C亚采样的最大还原因子覆盖深度为3.5x ~ 171x;对整个微生物群落的全基因组,使用last(v941)将SPADES集合产生的scaffolds与“封闭”的参考基因组对齐,构建出真实情况。
采用调整交互信息(AMI)(sklearnv0.19.2)和加权Bcubed(B3)两种方法验证基因组的分型,结果显示B3更准确;由于bin3C不针对组装校正,文章选择使用scaffolds而不是contigs做基因组拼接。使用BWA MEM (v0.7.17 r1188)将模拟的和真实的Hi-C读图映射到各自的scaffolds上。使用samtools (v1.9) 处理生成的BAM文件,以删除未映射的reads和补充及辅助对齐,然后按名称排序并合并。
所选63个基因组的ANI范围为74.8-95.8%(中位数77.1%),GC含量范围为28.3-73.8%(中位数44.1%)没有两个基因组的相似性超过96%, ANI限制了深度测序物种的过度表达(图1)。
那么在宏基因组测序深度不变的情况下,不同的Hi-C覆盖范围如何影响bin3C正确检索MAGs呢?
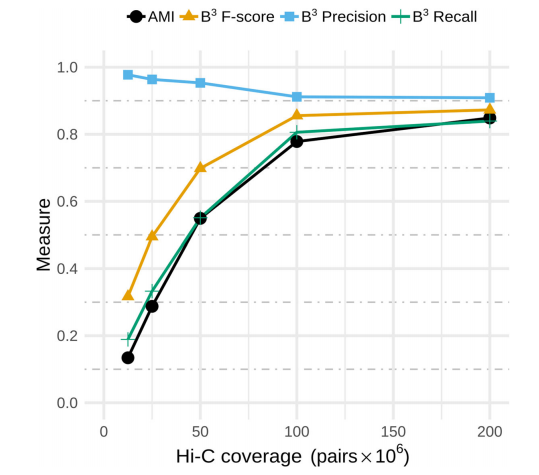
为了检验了bin3C的质量,Hi-C覆盖深度从12.5 M至200 M 互作(图2)。Hi-C覆盖范围从12.5 M增加到100 M互作时,AMI、B3和B3 F评分明显增加,而100 M和200 M对之间的增加变缓慢。而随着Hi-C覆盖度增加,B3精度降低,但是下降较少。
在200 M 互作时AMI、B3和B3 F达到最大值(0.848、0.839、0.873)。在此深度下大于1,000 bp的数据中,22,279个通过了bin3C过滤,占95.4%。共有62个基因组库大于50kbp,总大小为229,473,556bp,占整个宏基因组数据的95.6%,占参考基因组范围的91.1%。其余小于50kb范围的小集群总数为1,413,596 bp,占装配范围的0.6%,而低于1,000 bp未分析的为8,103,486 bp,占3.4%。
B3作为一种软聚类度量,既考虑了预测聚类内的重叠,又考虑了数据的真实性。在我们的模拟群体中,共享序列的区域为4.4%,意味着4.4%的序列分配是不明确的,由两个或多个源基因组共享。尽管如此,bin3C解决方案是硬集群,将重叠contigs放在一个bin中。即使没有错误,这也会使基本事实和最好的bin3C解决方案之间留下一个很小但无法逾越的鸿沟。
相反,AMI是一个硬聚类的方法,它需要通过抛硬币的过程将基本事实中的每个共享contigs分配给一个源基因组。然而,当bin3C为此类contigs选择一个bin时,任何一个源都同样有效。由于这个原因,AMI的分数在有重叠基因组的情况下不太可能实现统一。
尽管存在这些技术上的问题,但是当考虑到整个宏基因组组装的重复序列分配时,使用B3查全率和精密度对总体完整性和污染进行定量评估是可靠的。这与基于标记基因的完整性和污染的检测方法不同,只有那些包含标记基因的重叠基因组才会对检测结果产生影响。
随着Hi-C覆盖深度从12.5 M增加到200 M互作,bin3C的整体完备性从0.189增加到0.839。与此同时,用B3精密度推断的总体污染从0.977略微下降到0.909。因此,bin3C在保持总体低污染程度的同时,对Hi-C覆盖深度的增加做出了积极的响应。
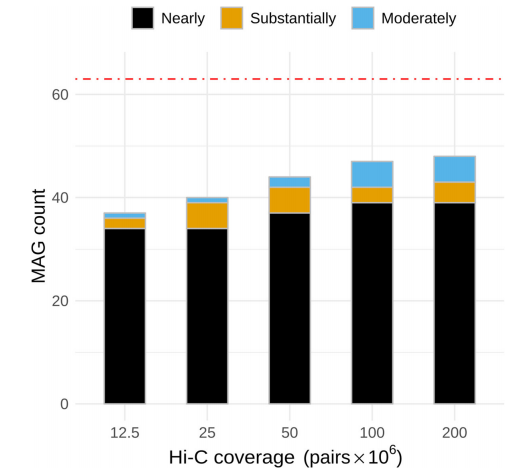
接着,使用标记基因工具CheckM验证了模拟菌群数据(图3)。对于相对较大的Hi-C深度覆盖范围的增加,检索到的MAGs数量的少量增加,为了解释这一点,我们参考了bin3C提供的聚类报告,其中对于接近完整的MAGs,我们发现平均数量的contigs从77对12.5 M pairs增加到179对200 M pairs,而contigs的总数从2,550对增加到6,968对。因此,尽管标记基因相关的叠盖层可以在较低的Hi-C覆盖深度有效地找到,但要获得每个MAG更完整的表达,需要更大的深度。
图3 使用CheckM验证使用bin3C检索的MAGs
由于bin3C既依赖于所提供数据的质量,又依赖于数据的数量,因此在Hi-C覆盖深度之外的这两个影响结果的因素进行处理是非常重要的。宏基因组测序数据是形成Hi-C关联的基础,因此,对一个群落的采样越彻底,效果就越好。
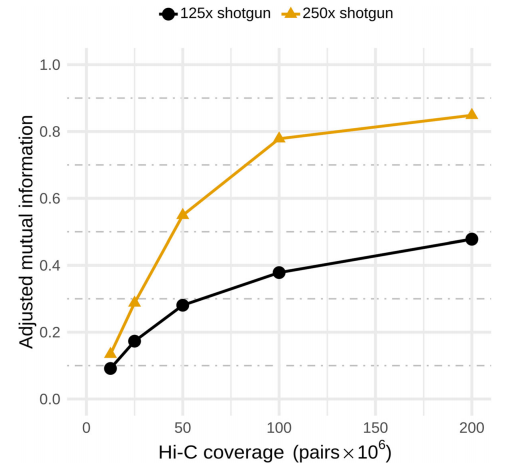
为了演示宏基因组数据对bin3C的影响,将模拟群落宏基因组测序深度降低了一半(至125x),并重新组装了宏基因组。这此深度下,组装数据为N50 6,289 bp和L50 4,353。长度超过1,000 bp的contigs共有43,712个,长度为187,388,993 bp,总数量为113,754个,长度为22,252,774 bp。这与全深度(250x)组装形成对比,全深度(250x)装配有N50 30,402 bp和L50 1,105, 23,364个contigs超过1,000 bp,总长度为232,030,334 bp,41,704个contigs,长度为240,133,820 bp。很明显,测序深度的降低导致了组装效果的下降。
然后,在宏基因组125X与250X时,分析了在相同的Hi-C覆盖深度范围内使用bin3C的效果,进行AMI验证得分的比对(图4)。二分之一深度集和全深度进行AMI验证得分的比对表明,对于采样更深入的群落bin3C对数据的组装提升更大。完整性和污染的CheckM遵循类似的趋势。
图4 两个不同宏基因组深度下bin 3C调整后的相互信息(AM)得分
在半深度的最佳结果生成了25个接近、4个基本和6个完全的MAGs模型,而在全深度的情况下生成了39个接近、4个基本和5个中等完整的MAGs。近年来,在制备宏基因组Hi-C文库的过程中,使用了两种不同的限制性酶。酶选择的再生位点有不同的GC偏差。对于一个物种多样的微生物群落,GC范围很广,这种策略的目的是更均匀地分离提取的DNA,从而覆盖整个宏基因组的Hi-C。
当基于Hi-C的进行基因组组装时,更均匀的覆盖会带来更好的结果。模拟一个双酶库,为了最接近真实实验。重新利用这个数据,以确定使用两个酶而不是单独使用一个酶获得了什么好处。模拟文库中使用的两种酶是Sau3Al和MluCL。虽然Sau3Al的限制位点^GATC是GC平衡的,但是MluCl的^AATT限制位点是AT富集的。模拟群落中,源基因组GC含量范围为28.3 ~ 73.8%,其丰度随机分布。对于Sau3AI,这些极端的GC含量转化为预期的每338个bp中有1个(28.3%)和每427个bp中有1个(73.8%)的剪切位点频率。对于不太平衡的MluCI,预期的切割频率为每61个bp中有1个,为28.3%,每3,396个bp中有1个,为73.8%。因此,MluCI的位点密度在低GC范围会非常高,而在高GC范围会非常稀疏。
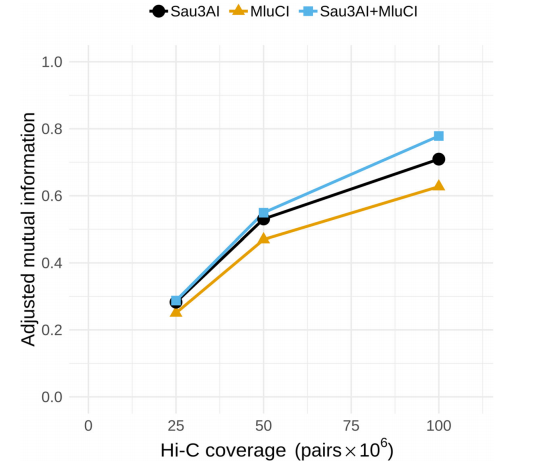
对于模拟的群落全深度组装,使用bin3C分析了三个Hi-C场景:使用Sau3Al或Sau3Al生成的两个单酶库和使用Sau3Al和MluCI的双酶文库。对相同Hi-C覆盖深度的库进行了性能评估。AMI而言,单一酶bin3C库的性能低于Sau3Al + MluCI相结合的文库(图5)。两种酶模型的优势增长随着深度的增加而增长,在Hi-C 100 M对互作时,AMI分数MluCI 0.63,Sau3Al 0.71而Sau3Al + MluCI 0.78。
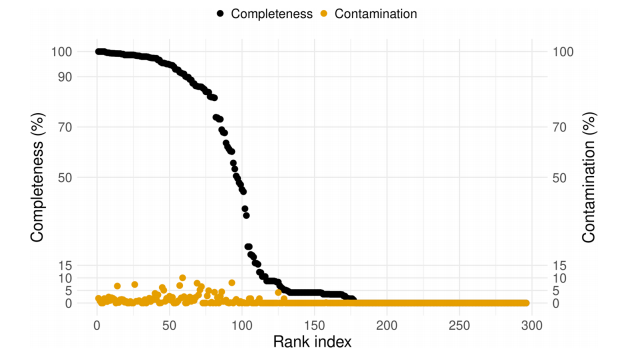
使用与模拟群落相同的参数,用bin3C分析了真实的人类肠道微生物组。在95,521个长度大于1,000 bp的contigs中,29,653个具有足够的信号被纳入聚类。大于1,000 bp的contigs的总长度为517,309,710 bp,而Hi-C足够的则为517,309,710 bp总观测值为339,181,288个基点,占总观测值的65.6%。超过50 kbp的296个簇的总长度为290,643,239 bp。聚类长度在10 kbp以上的为324,223,887 bp,占总聚类长度的45.1%。
图6 bin 3C从真实人体肠道菌群中检索MAGs,按完整性递减估计(黑色圆圈)排序
使用CheckM分析了这296个基因组(图6)。bin3C检索了近55个、基本29个和中等完整的12个MAGs。就整个范围而言。MAGs排名接近完成的范围为1.68 Mbp到4.97 Mbp,而基本完成的范围为1.56到5.46 Mbp,中等完成的范围为1.22到3.40 Mbp。在宏基因组覆盖范围方面,MAGs排名接近完整,从5.9x到447.5x,大致从4.3倍到416.4倍,适度从3.7倍到83.4倍。bin3C解决方案得到17个高质量、78个中等质量和105个低质量的MAGs。
使用bin3C分析的真实微生物组在之前的研究中首次被报道,是以演示一种称为ProxiMeta的宏基因组Hi-C分析服务。基于Hi-C的宏基因组组装,ProxiMeta是唯一的另一个完整的解决方案。由于ProxiMeta是一种专有服务,而不是开源软件,所以通过重新分析他们工作中使用的相同数据集进行了比较。
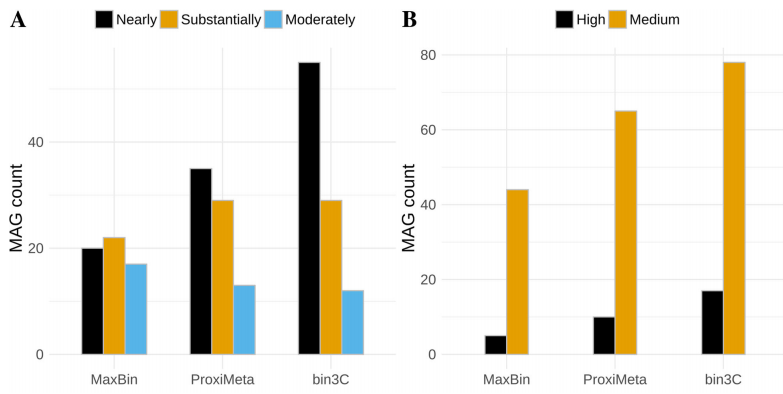
据报道ProxiMeta检索到35个接近、29个基本和13个中等完整的MAGs,而MaxBin检索到20个接近、22个基本和17个近似完整的MAGs。在相同的元基因组Hi-C数据集上,我们发现bin3C检索到55个接近、29个基本,和12个中等完成的MAGs(图7a)。
图7 MaxBin、ProxiMeta、bin3C在相同数据中检索到的MAGs
相对于MaxBin, bin3C检索到较少的中等完整的MAGs,但在其他方面显示它更高的性能。相对于ProxiMeta,bin3C在相当程度上和中等程度完成的序列中具有相同的性能,同时检索了另外20个接近完整的基因组,结果显示改进了57%。
结果表明,与MaxBin、ProxiMeta 相比,bin3C具有更高的组装精度,从而大大降低了污染率。当bin3C提升最高质量级别的MAGs时,主要是由于减少了对过量污染的回收。对于所有超过1 Mbp的基因组bin, bin3C的中位污染率为0.8%,而ProxiMeta中位污染为3.5%,MaxBin为9.5%。
结论
① bin3C这种公开的通用算法,可重复有效地检索模拟和真实宏基因组数据中的MAGs;
② 更高深度的宏基因组测序对MAGs检索的精确度和完整性有很强影响;
③ 与之前的MaxBin、ProxiMeta 相比,bin3C大大降低了污染率;
④ bin3C与MaxBin、ProxiMeta相比在人类肠道微生物组装中获得了更多完整的基因组;
⑤ 为获得最佳的结果,建议使用双酶消化模型构建Hi-C宏基因组文库;
⑥ bin3C可以分析小于1,000bp的序列,但它们进入分析并不能改善MAG检索。
配图来源网络/侵删
参考文献:
Matthew ZD , Aaron ED . bin3C: exploiting Hi-C sequencing data to accurately resolve metagenome-assembled genomes. Genome Biology. 2019.02.
 027-87224696
|
027-87224696
| marketing@frasergen.com
|
marketing@frasergen.com
|


 微信公众号
微信公众号

